CHECKALIGN 2.0
CheckAlign 2.0 computes two variations of the Shannon algorithm.
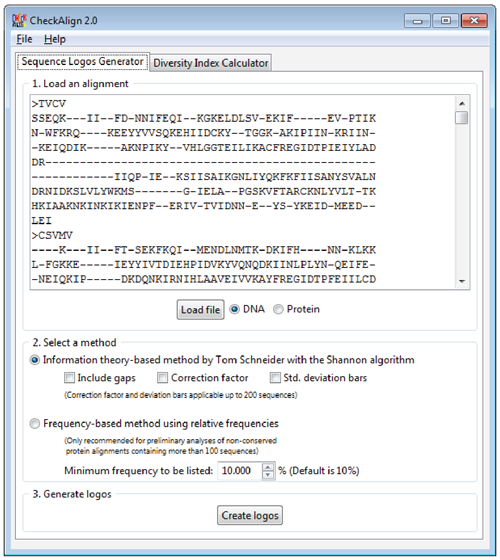
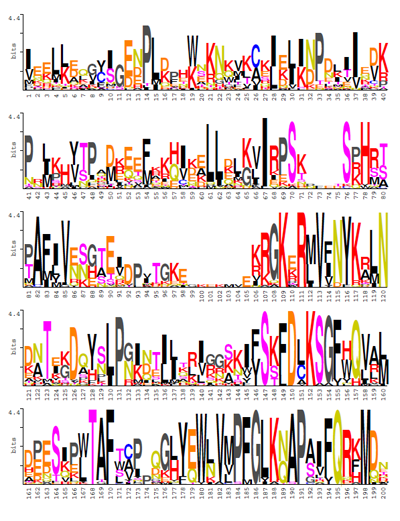
The tool computes the canonical algorithm introduced by Schneider and Stephens in previous logos methodologies approaches for constructing sequence logos from the input of gapped and ungapped protein and DNA multiple alignments. The resulting logos represent each column of a sequence alignment as a stack of letters of variable color and height depending on the amino acid or nucleotide chemical nature and frequency.
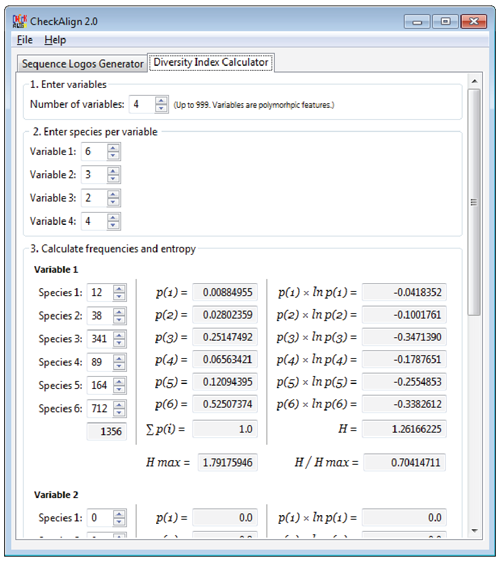
CheckAlign 2.0 also focuses on other aspects of information theory focusing concerning the diversity of species in an ecological community. In this case, the tool provides accounts of both the abundance and evenness of the species present in a community sample, by computing the Shannon-Weaver diversity index (H) and the Equitability (EH) of these samples.
CheckAlign 2.0 is distributed as a standalone software adapted to the most common operating systems or as an online server accessible at the Gypsy Database of Mobile Genetic Elements.
FEATURES
Free access to the tool
New implementations focused on quantification of population diversity
Adjusted to the new versions and updates of the most common operating systems
Open source application available both as standalone software and as an online server
CheckAlign 2.0 is a Java application that runs on the most common operative systems
ABOUT CHECKALIGN
Information theory is a branch of applied mathematics involving the quantification of information. Soon after its inception, information theory was applied to distinct areas of biological research. Of significant interest was the sequence logo methodology developed by Schneider and Stephens in 1990, which offers graphical representations of DNA or protein multiple alignments, providing a statistically relevant visualization of the consensus of a set of sequences, their common information content, and the frequency of all possible DNA and amino acid states per alignment position.
The new version CheckAling 2.0 includes improvements and updates devoted to both source code optimization and statistical analysis enhancement. Users can use CheckAlign 2.0 to build logos online and/or on their PCs using the conventional methodology of Schneider and Stephens where the application is especially useful to decipher the order of predominance and relative frequencies of residues at every position, providing a significant view of the patterns that characterize the architecture of a gene or a protein.
Users can also use CheckAlign 2.0 to apply other information theory principles in order to characterize the rarity and commonness of species in a biological or molecular community